总结在RiboMiner使用,RiboMiner功能太多了,对不上号,总结一下。
1. metaplot
bam文件需要sort并建立index。
1 | samtools index -@ 10 01.bam/my_file.mapped.sorted.bam; |
得到的图: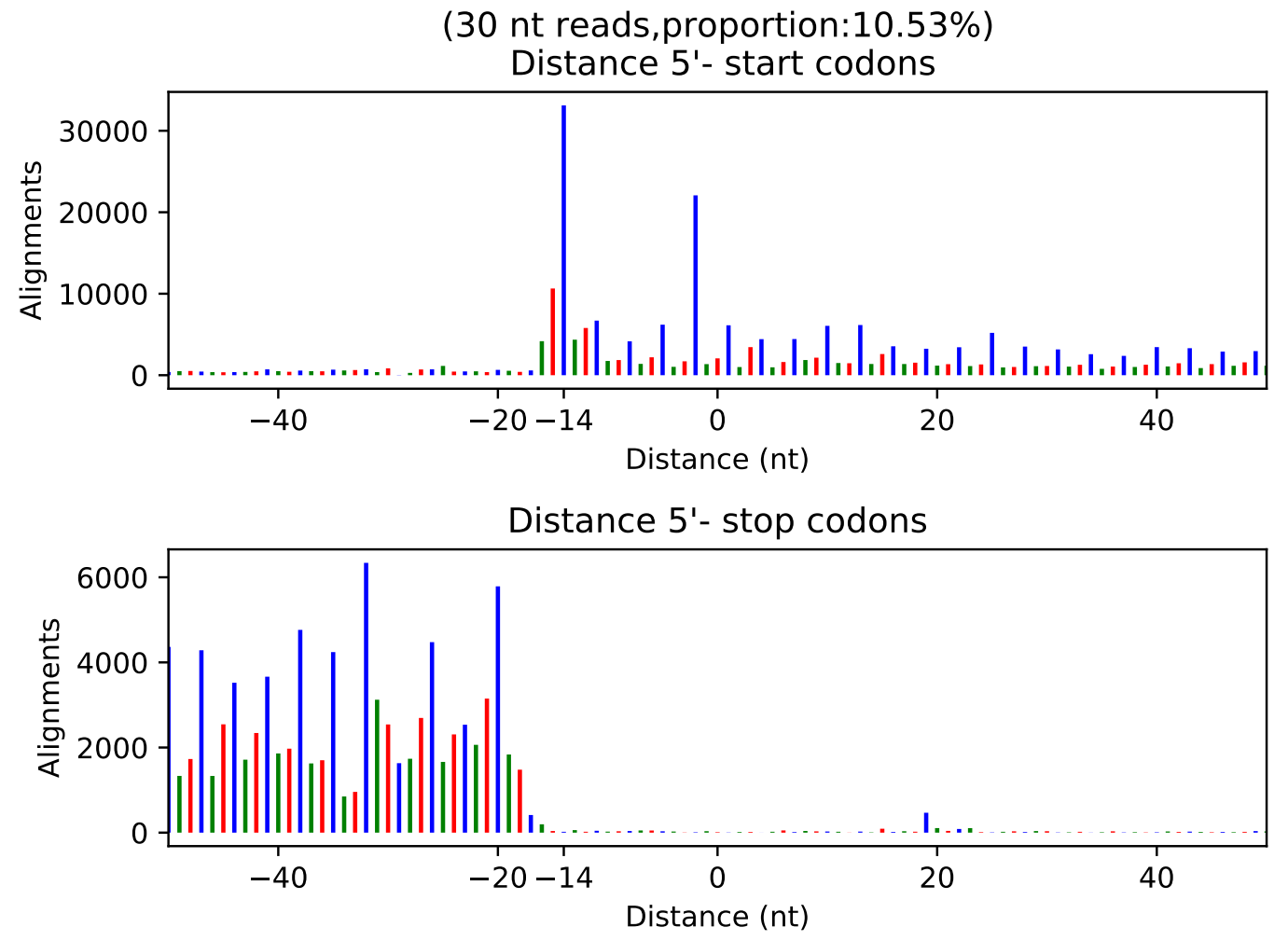
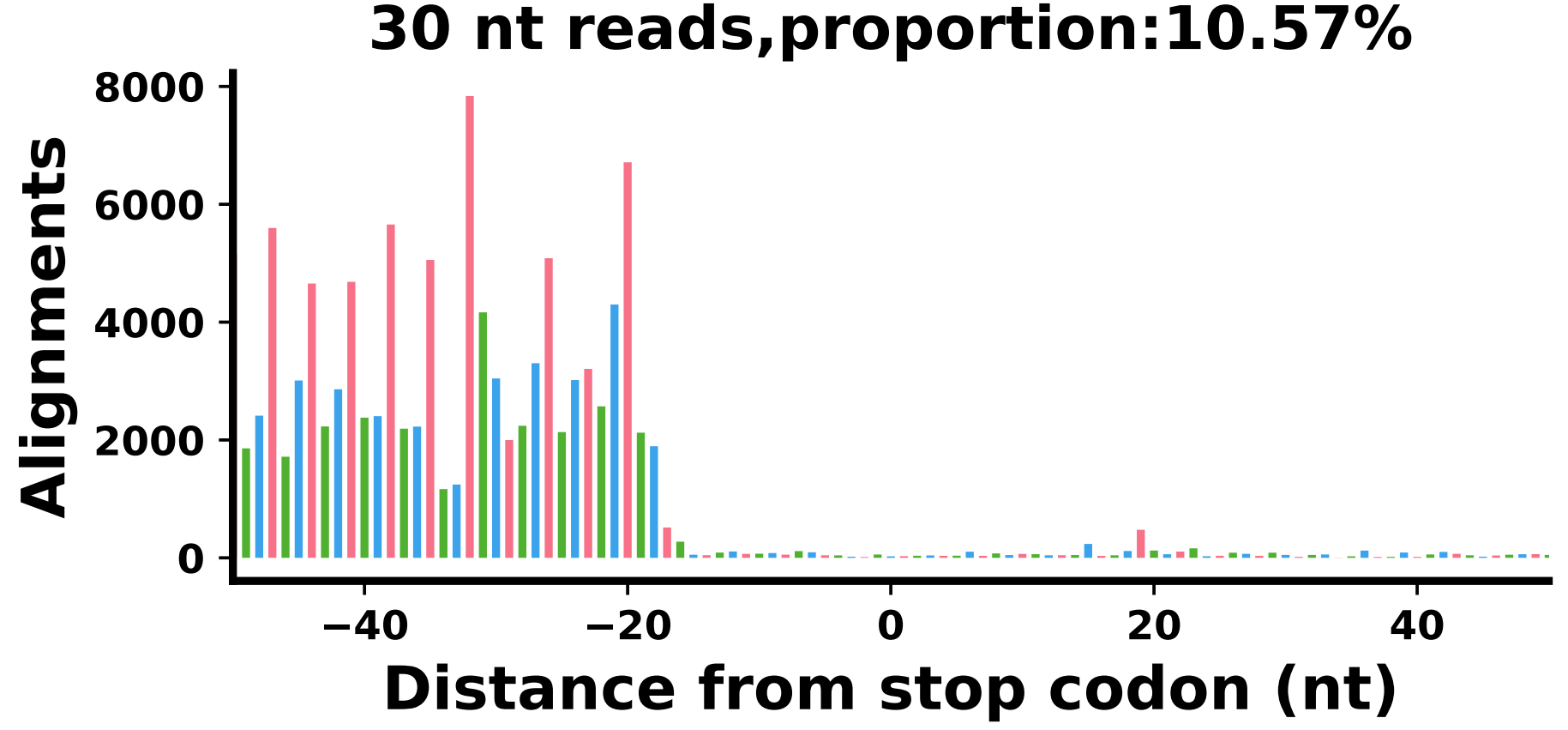
2. periodicity
1 | Periodicity -i 01.bam/my_file.mapped.sorted.bam -a Ribo_ann/ -o 02.Periodicity/my_file -c Ribo_anno/longest.transcripts.info.txt -L 25 -R 35; |
得到的图: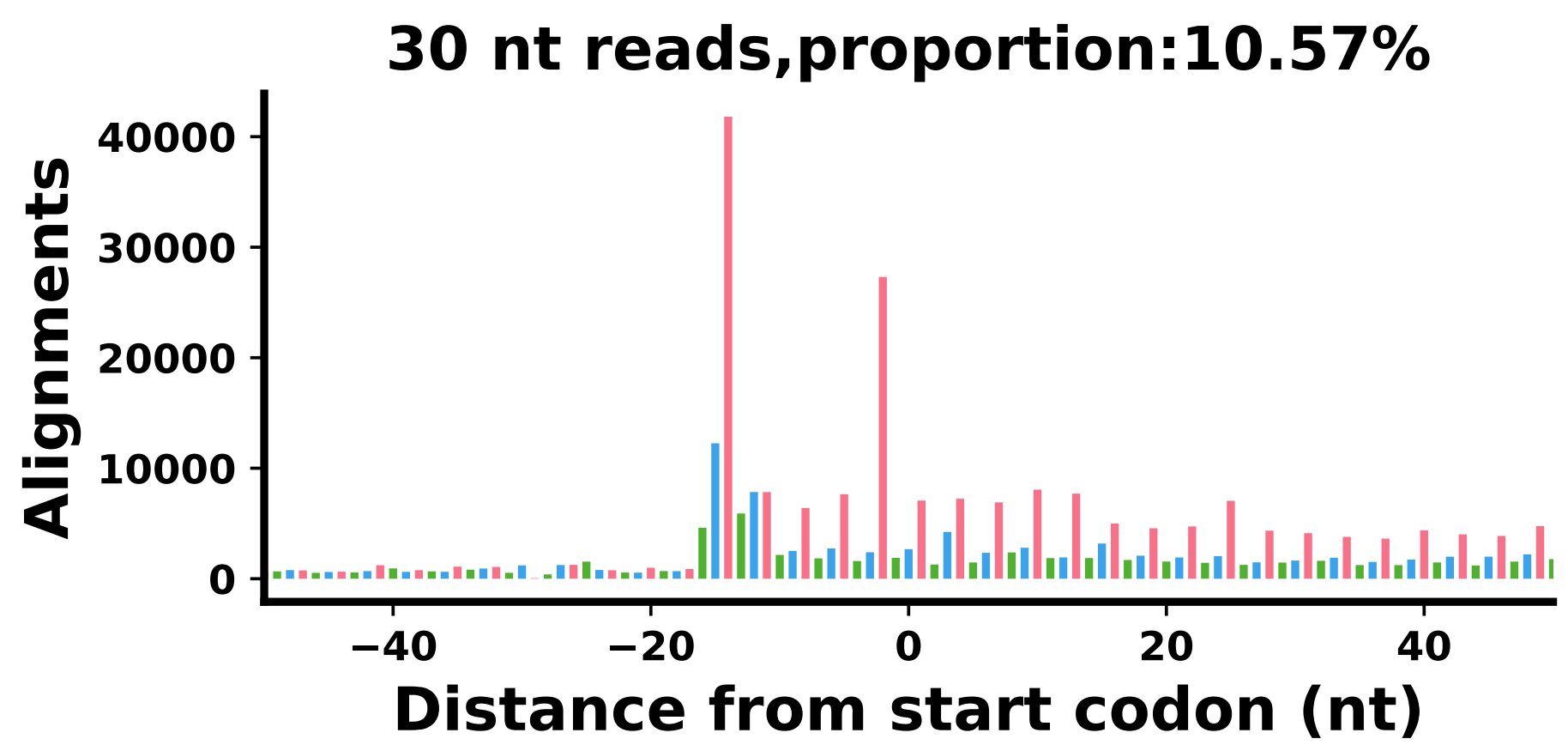
3. lengthdistribution
1 | LengthDistribution -i 01.bam/${i}.mapped.sorted.bam -o 03.ReadLength/${i} -f bam; |
得到的图: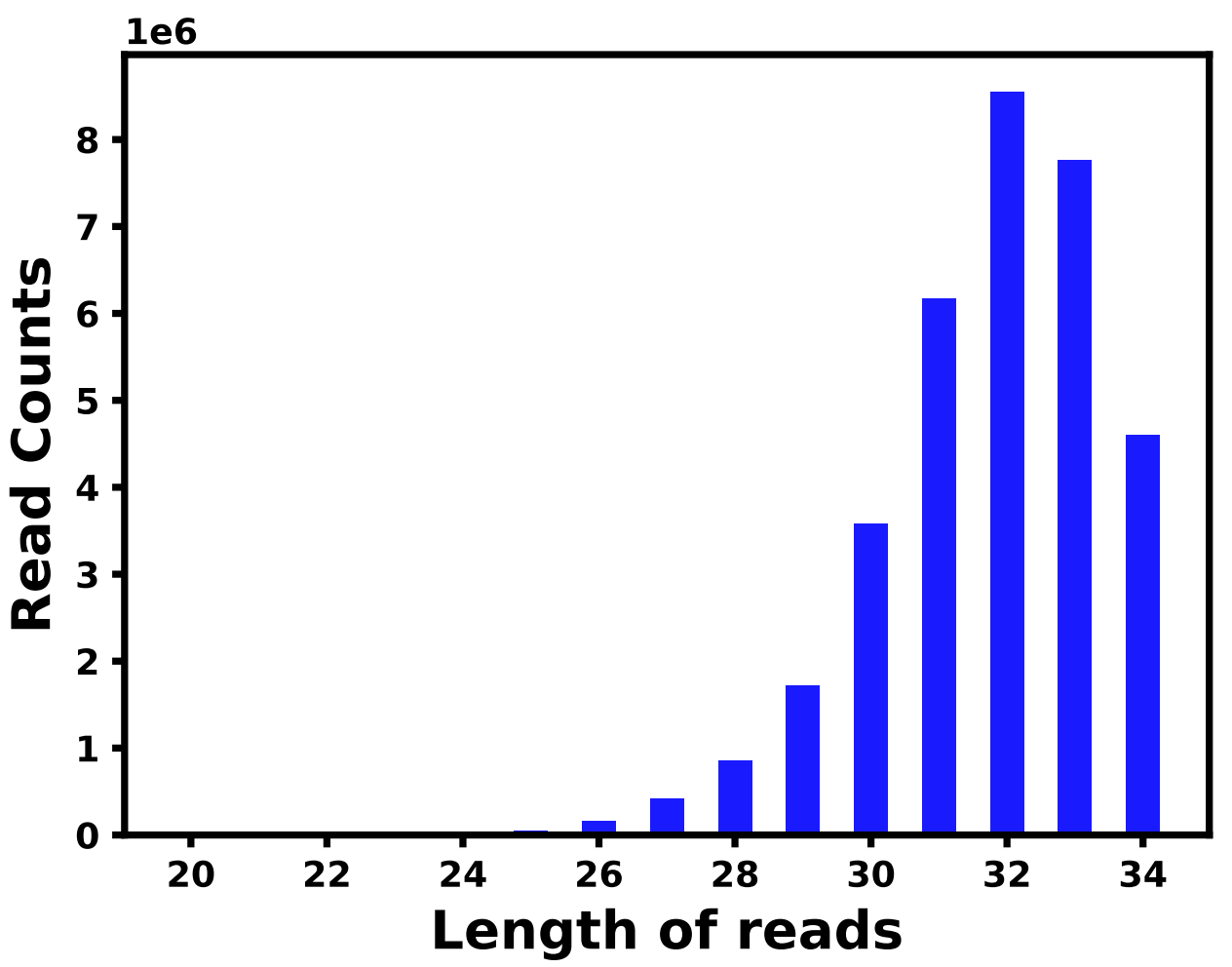
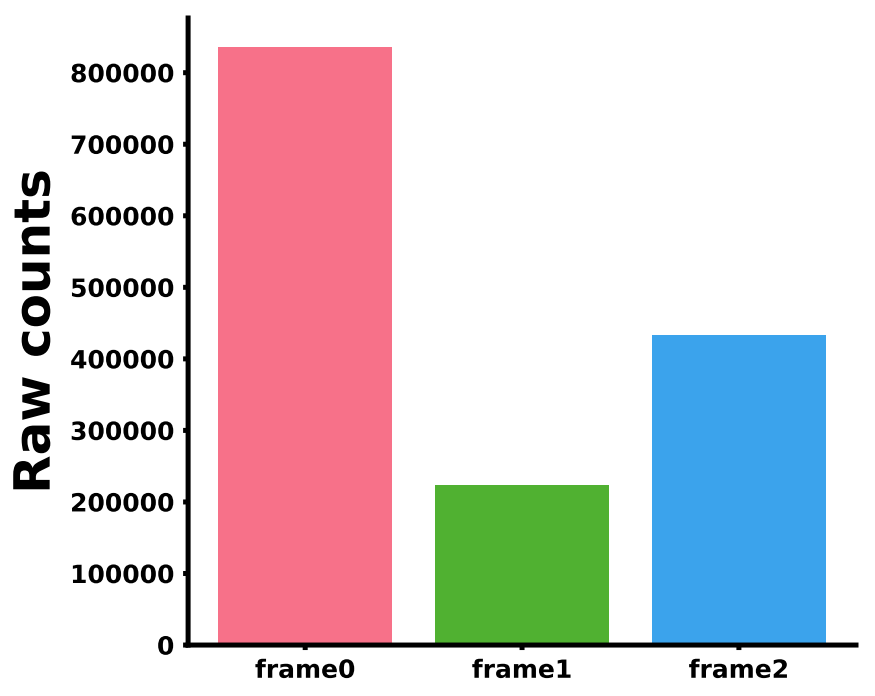
4. RiboDensityOfDiffFrames
attributes.txt
1 | bamFiles readLengths Offsets bamLegends |
里面有几个文件,针对每一个文件都会单独产生一个图。
1 | RiboDensityOfDiffFrames -f attributes.txt -c Ribo_anno/longest.transcripts.info.txt -o 04.RiboDensityofDiffFrames/ {-S select_trans.txt --id-type transcript-id --plot yes}; |
得到的每个bam文件的图如下: